

Developing medical devices for the global market is a complex process, not only because of technical and clinical requirements but also due to the strict regulatory frameworks that govern product safety, effectiveness, and data protection. Compliance with regulations is a critical factor for successfully bringing a product to market, whether in the US or the European Union (EU). Medical device developers face a range of challenges, from understanding FDA regulations and EU MDR 2021 standards to implementing international quality management systems like ISO 13485:2016 and risk management frameworks such as ISO 14971:2019.
The consequences of failing to meet regulatory requirements can be severe: delays in product launch, costly redesigns, legal penalties, or even market withdrawal. According to industry studies, a significant percentage of medical devices fail regulatory approval due to incomplete documentation, inadequate risk management, or insufficient quality control processes. Addressing these challenges requires a strategic approach to regulatory compliance, integrated early into the product development lifecycle.
In this post, we’ll explore the core regulatory differences between the US and EU markets and how compliance directly shapes product design, approval, and launch strategies.
- Why Compliance Matters in US and EU Markets
- Overview of US and EU Regulatory Frameworks
- Key Standards and Regulatory Requirements
- Common Reasons Products Fail Regulatory Oversight
- Comparative Strategies for US vs EU Compliance
- Practical Recommendations for Development Teams
- Strategic Cost Analysis: Budget Planning for Market Entry
- Final Thoughts
Why Compliance Matters in US and EU Markets
In the United States, medical devices are regulated by the FDA (Food and Drug Administration). The FDA enforces regulations such as 21 CFR 820 for quality systems and CFR Part 11 for electronic records and signatures, ensuring that products meet strict safety and efficacy standards. For devices that handle Protected Health Information (PHI), adherence to HIPAA regulations is mandatory, covering data encryption, secure hosting, access control, and audit logging. Learn more about FDA regulations here.
In the European Union, regulatory oversight is governed by the EU Medical Device Regulation (MDR 2017/745), which replaced the previous Medical Device Directive (MDD) in 2021. The EU MDR emphasizes comprehensive clinical evaluation, post-market surveillance, and robust risk management, aligning closely with ISO 14971 standards. Manufacturers must demonstrate compliance through detailed technical documentation, continuous monitoring, and adherence to harmonized standards like ISO 13485. EU MDR details can be found here.
Medical device developers must navigate not only the legal requirements but also the technical and procedural challenges of maintaining quality throughout the product lifecycle. Failure to integrate ISO standards, risk management practices, and data security measures into the development process can result in regulatory setbacks, delayed market entry, or costly non-compliance penalties.
Impact of Regulations on Product Development and Market Entry
Regulatory frameworks influence nearly every aspect of medical device development:
- Design and development procedures must be meticulously documented and maintained.
- Software development for medical devices must comply with standards such as IEC 62304:2006, ensuring lifecycle control and patient safety.
- Agile practices can be implemented under guidance like AAMI TIR45, but teams must balance speed with rigorous documentation and traceability.
- Post-market surveillance and CAPA (Corrective and Preventive Actions) systems must be in place to continually verify compliance and manage emerging risks.
Understanding and addressing these regulatory challenges early not only accelerates time-to-market but also builds trust with regulators, clinicians, and patients. A strategic, compliance-first approach ensures that medical devices are both safe and market-ready, positioning companies for long-term success in both US and EU markets.
Overview of US and EU Regulatory Frameworks
Navigating the regulatory landscape for medical devices requires a clear understanding of the distinct approaches taken by the United States and the European Union. While both regions aim to ensure patient safety, product efficacy, and data protection, their frameworks differ in structure, enforcement, and compliance processes.
Key Regulatory Bodies: FDA vs EU MDR
In the United States, the Food and Drug Administration (FDA) is the primary regulatory authority for medical devices. The FDA classifies devices into three classes (I, II, III) based on risk level, with each class subject to different regulatory requirements. Compliance involves adherence to 21 CFR 820 for quality systems, CFR Part 11 for electronic records, and HIPAA regulations for protecting Protected Health Information (PHI). Devices handling patient data must ensure secure data storage, encryption, and audit trails. FDA guidance for medical devices can be accessed here.
In the European Union, regulatory oversight is governed by the EU Medical Device Regulation (MDR 2017/745), which replaced the previous Medical Device Directive (MDD) in 2021. The EU MDR introduces stricter requirements for clinical evaluation, post-market surveillance, and risk management. Manufacturers must maintain comprehensive technical documentation, perform conformity assessments, and adhere to harmonized standards such as ISO 13485:2016 for quality management and ISO 14971:2019 for risk management. EU MDR official documentation is available here.
Differences in Documentation, Risk Management, and Audits
Documentation Requirements
- US: The FDA requires thorough documentation of design controls, manufacturing processes, testing results, and CAPA actions. Records must be verifiable and easily retrievable for inspections.
- EU: The EU MDR mandates a Technical File or Design Dossier containing device specifications, clinical evaluation reports, risk management files, and post-market surveillance plans. Compliance with ISO standards is often used to demonstrate conformity.
Risk Management
- US: The FDA expects a structured risk management plan, often aligned with ISO 14971 principles, to identify, evaluate, and mitigate potential hazards.
- EU: EU MDR emphasizes continuous risk assessment throughout the device lifecycle. Risk management must feed into clinical evaluation and post-market monitoring.
Audits and Inspections
- US: FDA inspections can occur at manufacturing sites, development labs, or during post-market surveillance. Companies must demonstrate adherence to Quality System Regulations (QSR) and CAPA procedures.
- EU: Notified Bodies conduct audits to verify MDR compliance, including reviewing technical documentation, risk files, and quality management systems. Post-market surveillance reports must be regularly submitted.
Why Understanding Both Frameworks Matters
For companies aiming to market medical devices globally, awareness of these differences is crucial:
- A device developed solely to meet FDA standards may still fail EU MDR audits without additional documentation or risk management processes.
- Early integration of both ISO standards and regional regulatory requirements reduces time-to-market and avoids costly redesigns or delays.
- Combining agile development practices under frameworks like AAMI TIR45 with rigorous compliance ensures that software updates or iterations remain within regulatory boundaries.
By understanding and strategically addressing both US and EU regulatory frameworks, medical device developers can streamline product development, reduce compliance risks, and position themselves for success in both markets.
Comparison of US vs EU Regulatory Frameworks for Medical Devices
| Aspect | United States (FDA) | European Union (EU MDR) | Notes |
| Regulatory Authority | FDA (Food and Drug Administration) | EU Notified Bodies under EU MDR 2017/745 | EU MDR replaced MDD in 2021 |
| Device Classification | Class I, II, III based on risk | Class I, IIa, IIb, III based on risk | EU classification is slightly more granular |
| Quality Management Standard | 21 CFR 820 (QSR) | ISO 13485:2016 | Both require robust quality systems |
| Risk Management | Risk management aligned with ISO 14971 | Continuous risk assessment, ISO 14971 compliance recommended | EU emphasizes post-market risk monitoring |
| Electronic Records & Signatures | CFR Part 11 | MDR allows electronic documentation; compliance with EU data protection required | FDA has explicit rules for e-records |
| Data Protection | HIPAA for PHI, secure hosting, encryption, audit logs | GDPR for personal data; MDR emphasizes data protection in clinical evaluation | HIPAA is US-specific, GDPR applies in EU |
| Clinical Evidence | Required based on device class; submission for premarket approval (PMA) if Class III | Comprehensive clinical evaluation mandatory for all higher-risk devices | EU MDR often requires more detailed documentation |
| Post-Market Surveillance | FDA requires reporting of adverse events, recalls, CAPA | EU MDR requires periodic safety update reports, vigilance reporting | Both emphasize CAPA but EU MDR integrates lifecycle monitoring |
| Audits / Inspections | FDA inspections at manufacturing sites, development labs, or post-market | Notified Body audits of Technical File / Design Dossier and quality system | Audits are mandatory in both, but process differs |
| Software Requirements | IEC 62304 guidance for medical device software | IEC 62304 recommended, integrated with risk management | Software lifecycle standards apply in both regions |
| Agile Practices | Allowed, but must meet documentation & traceability requirements (AAMI TIR45) | Allowed under MDR if traceability & risk documentation maintained | Agile is feasible but must align with compliance |
Key Standards and Regulatory Requirements
Successfully navigating medical device regulations requires a deep understanding of both international standards and regional regulatory requirements. Compliance is not just a legal necessity – it ensures patient safety, product reliability, and market readiness. This section provides a comprehensive overview of the key standards and requirements that guide medical device development in the US and EU markets.
International Standards as a Foundation
ISO 13485:2016 – Quality Management for Medical Devices
ISO 13485 sets the framework for a robust quality management system (QMS) in medical device development. It ensures that every stage of the product lifecycle – from design and development to production, installation, and servicing – adheres to strict quality standards. Compliance with ISO 13485 is recognized globally and serves as a foundation for both FDA and EU MDR audits.
ISO 14971:2019 – Risk Management
Effective risk management is critical to patient safety. ISO 14971 provides a structured approach to identify, evaluate, and mitigate risks throughout the device lifecycle. Regulatory authorities expect documented evidence that risks have been systematically assessed, prioritized, and controlled. Integrating ISO 14971 into your development process helps ensure compliance with both FDA and EU MDR requirements.
IEC 62304:2006 – Medical Device Software Lifecycle
Software-driven devices are subject to specific regulatory scrutiny. IEC 62304 defines life-cycle requirements for medical device software, covering development, maintenance, and risk management. Compliance ensures that software is safe, reliable, and aligned with device risk classifications.
AAMI TIR45 – Agile Practices in Regulated Development
Agile development can accelerate product delivery, but medical device teams must ensure traceability and regulatory compliance. AAMI TIR45 provides guidance on how to implement Agile methodologies without compromising documentation, risk management, or quality standards.
US Regulatory Requirements
FDA Regulations – 21 CFR 820 & CFR Part 11
- 21 CFR 820: Governs the Quality System Regulation (QSR) for medical devices, covering design controls, production processes, CAPA, and documentation requirements.
- CFR Part 11: Ensures trustworthy electronic records and signatures, which is critical for audit readiness and compliance verification.
HIPAA Compliance
For devices handling Protected Health Information (PHI), HIPAA compliance is mandatory. Key considerations include:
- Hosting – servers must be HIPAA-compliant, with Business Associate Agreements (BAA) signed.
- Data Protection – PHI must be encrypted in databases and during transfer using SSL/TLS protocols.
- Backups & Disaster Recovery – encrypted backups stored in multiple locations; recovery strategies must be tested.
- Development Security – Dev and Prod environments must be separated; sensitive data must be masked or anonymized.
- Access Control – implement role-based access (RBAC) and secure authentication, including strong passwords and two-factor authentication (2FA).
- Image & Mobile App Protection – sensitive imaging data (e.g., MRI scans) must not be cached; mobile apps must ensure secure storage and access.
EU Regulatory Requirements
EU Medical Device Regulation (MDR 2017/745)
The EU MDR sets comprehensive requirements for clinical evaluation, post-market surveillance, and technical documentation. Compliance involves:
- Maintaining a Technical File or Design Dossier that documents device specifications, clinical data, risk assessments, and CAPA procedures.
- Aligning quality management systems with ISO 13485.
- Continuous risk management and post-market surveillance, demonstrating that devices remain safe and effective throughout their lifecycle.
Key Takeaways for Development Teams
- Integrate ISO standards early into the development process.
- Ensure FDA and EU MDR compliance is considered in design, testing, and documentation.
- Apply Agile development practices following AAMI TIR45 to maintain regulatory alignment.
- Prioritize HIPAA and GDPR compliance for all devices handling sensitive patient data.
- Establish robust CAPA systems, maintain traceable documentation, and prepare for audits in both regions.
By adhering to these standards and regulatory requirements, medical device teams can reduce the risk of regulatory failure, accelerate time-to-market, and ensure that devices are safe, effective, and trusted by clinicians and patients alike.
Common Reasons Products Fail Regulatory Oversight
Medical devices often fail regulatory review not because of poor engineering, but due to gaps in compliance, documentation, and quality management. Understanding these pitfalls can help development teams anticipate challenges, implement proactive measures, and significantly reduce the risk of delays, recalls, or rejection.
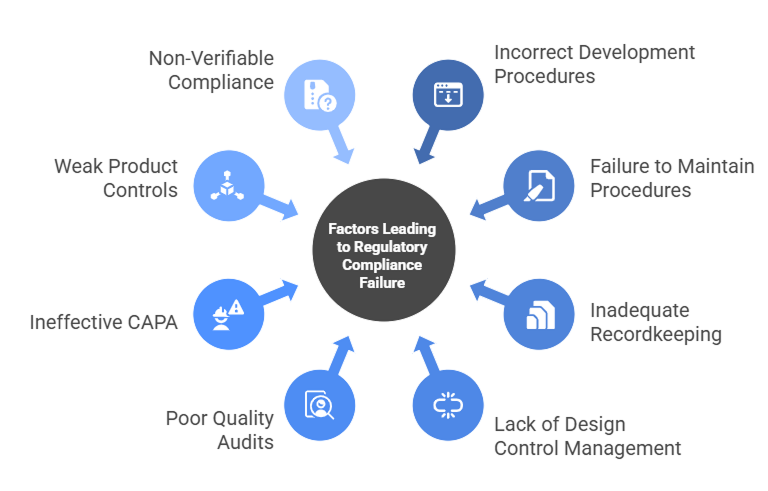
1. Incorrectly Established Development Procedures
Regulators such as the FDA and EU MDR authorities expect well-defined and structured development procedures. Without clear processes for design, testing, risk assessment, and verification, teams may produce inconsistent results.
Example: A software-driven medical device team skips documenting specific validation steps for a new firmware update. During FDA inspection, auditors cannot verify that testing aligns with IEC 62304 lifecycle requirements, resulting in non-compliance findings.
Insight: Early investment in standardized procedures, including flowcharts, checklists, and templates, helps ensure repeatability and regulatory readiness.
2. Failure to Maintain Procedures
Even well-defined procedures can become obsolete. Standards and regulations are updated frequently – ISO 13485:2016, ISO 14971:2019, and EU MDR 2021 all include revisions that impact documentation, risk management, and clinical evidence.
Example: A company continues using legacy MDD templates after the MDR came into force, ignoring updated requirements for post-market surveillance and risk management. This leads to audit findings and delayed certification.
Insight: Maintain a living compliance program – regularly review procedures, train staff on updates, and revise documentation accordingly.
3. Inadequate Recordkeeping
Regulators demand that every step of device development is documented, traceable, and verifiable. This includes design inputs, risk analyses, verification and validation results, and CAPA records.
Example: Risk assessment notes are stored only in personal files rather than a central repository. When an EU MDR auditor requests evidence of hazard mitigation per ISO 14971, the team cannot produce verifiable records.
Insight: Implement a centralized document management system to capture all records in a structured, auditable format.
4. Lack of Design Control Management
Design control ensures that device specifications, design outputs, and verification processes align with regulatory expectations. Missing traceability between requirements and outputs is a frequent reason for rejection.
Example: A Class II device is released without documented verification that software updates meet initial safety requirements. Auditors cite the missing link as a critical gap in ISO 13485 QMS compliance.
Insight: Use traceability matrices to link design inputs to outputs, risk mitigations, testing results, and CAPA actions. This ensures auditors can quickly verify compliance.
5. Poor Quality Audits
Regular internal audits are essential to identify deviations before external inspections. Weak or inconsistent audits allow minor issues to accumulate into major compliance failures.
Example: During a routine FDA inspection, a device fails because prior internal audits did not flag recurring software bugs in the mobile app, which impact HIPAA compliance for PHI.
Insight: Establish a robust audit schedule that includes cross-functional reviews of design, software, risk management, and CAPA systems.
6. Ineffective Corrective and Preventive Actions (CAPA)
CAPA is the backbone of continuous improvement and compliance verification. Ineffective CAPA systems allow problems to recur, raising regulatory concerns.
Example: A manufacturer notices recurring errors in device labeling. CAPA records indicate the issue was “resolved” without root cause analysis. During EU MDR assessment, auditors reject the solution as inadequate.
Insight: CAPA must include root cause analysis, documented corrective measures, and preventive strategies, with evidence that actions are effective.
7. Weak Product Controls
Product controls – specifications, testing protocols, and release criteria – must be rigorously applied. Missing or inconsistent controls lead to unpredictable product quality.
Example: A connected medical device’s firmware lacks version control, resulting in multiple devices running untested software in the field. FDA inspectors flag this as a serious compliance risk.
Insight: Define clear release criteria, implement version control for software and firmware, and ensure all product variations meet documented standards.
8. Non-Verifiable Compliance
Finally, compliance must be documented, verifiable, and auditable. Regulators must trace every decision, test result, and CAPA action to ensure safety and conformity with standards like ISO 13485, IEC 62304, FDA QSR, or EU MDR.
Example: Clinical evaluation reports lack links to supporting risk files. EU MDR auditors cannot verify that all risks were mitigated, resulting in non-conformity.
Insight: Use integrated compliance tools and centralized repositories to make all regulatory evidence traceable and verifiable at any time.
Comparative Strategies for US vs EU Compliance
Navigating both US and EU regulatory frameworks simultaneously can be challenging. Each market has distinct requirements for documentation, risk management, quality systems, and data protection. However, strategic planning allows development teams to align processes efficiently, reduce duplication of effort, and ensure global compliance.
1. Documentation and Record-Keeping
US (FDA):
- Requires comprehensive records for design controls, production, CAPA, and electronic data (CFR Part 11).
- Documentation must be auditable, verifiable, and retained for inspection.
EU (MDR 2017/745):
- Emphasizes a Technical File or Design Dossier containing device specifications, clinical data, risk assessments, and post-market surveillance plans.
- Documents must demonstrate conformity to ISO 13485 and ISO 14971.
Strategy:
- Implement a centralized document management system covering both FDA and EU requirements.
- Use templates aligned with ISO standards to ensure consistency.
- Maintain version control and audit trails to satisfy both regulatory authorities.
2. Risk Management Approaches
US: FDA expects a structured risk management plan, typically aligned with ISO 14971, to assess, mitigate, and document potential hazards.
EU: EU MDR mandates continuous risk assessment throughout the device lifecycle, integrating risk into clinical evaluation and post-market surveillance.
Strategy:
- Develop a unified risk management framework that meets both FDA and MDR expectations.
- Maintain traceability from risk identification to mitigation, verification, and documentation.
- Regularly update risk files based on post-market feedback or software changes.
3. Data Security Standards
US:
- HIPAA compliance is mandatory for PHI, including secure hosting, encryption, role-based access, and audit logs.
- Software and mobile applications must safeguard sensitive patient data throughout development and production.
EU:
- GDPR governs personal data protection, including medical information used in clinical evaluation and device operation.
- MDR emphasizes secure handling of clinical data and post-market reporting.
Strategy:
- Implement data protection protocols that meet both HIPAA and GDPR requirements.
- Separate development and production environments to prevent unauthorized access to real patient data.
- Encrypt sensitive data at rest and in transit, and log all access events for audit purposes.
4. Software and Agile Development
US:
- FDA supports Agile development under guidance like AAMI TIR45, provided documentation and traceability requirements are met.
- Software must comply with IEC 62304 lifecycle standards.
EU:
- MDR allows Agile, but documentation, traceability, and post-market surveillance must be maintained.
- IEC 62304 is recommended for software validation and risk integration.
Strategy:
- Use Agile practices with structured documentation to satisfy both FDA and EU MDR.
- Maintain traceable links from user stories and software features to risk mitigation and verification.
- Conduct regular internal audits to verify compliance during iterative development cycles.
5. CAPA and Quality Management
US: FDA requires an effective CAPA system integrated into the QMS (21 CFR 820).
EU: MDR emphasizes CAPA as part of continuous improvement, linked to post-market surveillance.
Strategy:
- Integrate CAPA into a single, centralized quality management system.
- Ensure corrective actions are documented, validated, and reviewed across both markets.
- Conduct periodic audits to ensure CAPA effectiveness and regulatory readiness.
Comparative Compliance Strategies: US (FDA) vs EU (MDR)
| Area | US (FDA) | EU (MDR 2017/745) | Strategic Alignment |
| Documentation & Record-Keeping | Requires detailed records for design controls, production, CAPA, and electronic data (21 CFR Part 11). Must be auditable and verifiable. | Focuses on Technical File/Design Dossier with specifications, risk analysis, clinical data, and PMS plans. Conformity to ISO 13485 & ISO 14971 required. | Use a centralized document management system; align templates with ISO standards; maintain version control and audit trails. |
| Risk Management | Structured plan aligned with ISO 14971; focuses on identifying, assessing, and mitigating hazards. | Continuous risk assessment across lifecycle, integrated with clinical evaluation and PMS. | Build a unified risk management framework meeting both standards; maintain traceability and update risk files regularly. |
| Data Security & Privacy | HIPAA compliance required for PHI — encryption, secure hosting, role-based access, and audit logs. | GDPR governs personal data and MDR enforces secure data handling and reporting. | Implement data protection protocols that satisfy both HIPAA & GDPR; encrypt data, control access, and maintain audit logs. |
| Software & Agile Development | FDA supports Agile (AAMI TIR45) if traceability and IEC 62304 compliance are maintained. | Agile permitted under MDR with strong documentation and post-market traceability. | Apply Agile with structured documentation; link user stories to risk and verification; perform internal audits. |
| CAPA & Quality Management | CAPA system required under 21 CFR 820; integral to QMS for corrective/preventive actions. | CAPA part of continuous improvement under MDR, tied to PMS feedback. | Centralize CAPA within the QMS; document and validate all actions; audit regularly to ensure compliance and readiness. |
Practical Recommendations for Development Teams
Successfully navigating the regulatory landscape for medical devices in the US and EU requires a proactive and systematic approach. Development teams can reduce delays, avoid costly rework, and improve approval success by integrating compliance into every stage of the product lifecycle. Here are actionable recommendations based on best practices and regulatory expectations.
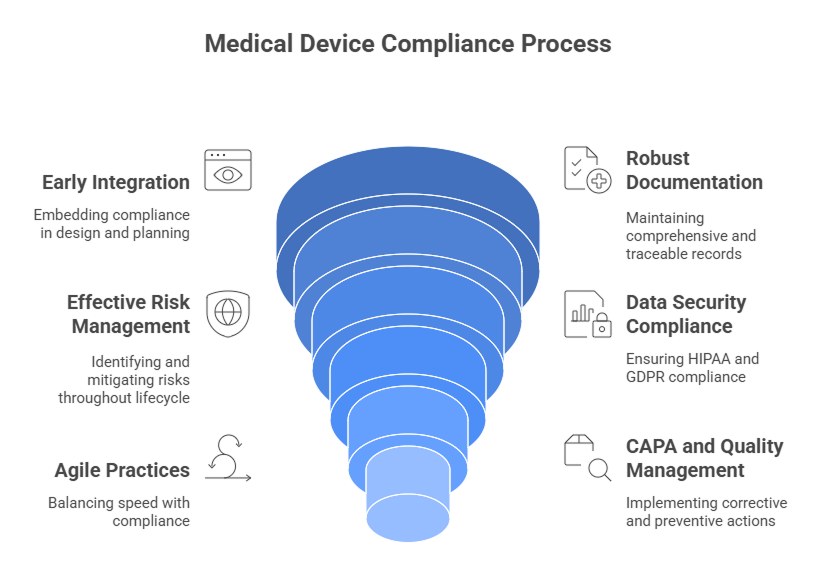
1. Integrate Regulatory Requirements Early
- Embed FDA, EU MDR, ISO 13485, and ISO 14971 requirements at the concept and design phases.
- Define clear design inputs and outputs, traceable to user needs, safety standards, and regulatory obligations.
- Plan for clinical evaluation, risk assessment, and data protection from the beginning to avoid last-minute adjustments.
Insight: Early compliance reduces the risk of audit findings and accelerates time-to-market.
2. Maintain Robust Documentation
- Centralize all records in a document management system to ensure traceability.
- Include design specifications, risk assessments, test results, CAPA actions, and clinical evidence.
- Ensure documentation aligns with ISO standards, FDA QSR, and EU MDR Technical File requirements.
Tip: Use checklists and templates to maintain consistency across teams and markets.
3. Implement Effective Risk Management
- Follow ISO 14971:2019 to identify, evaluate, and mitigate risks across the device lifecycle.
- Integrate risk assessment into software development and firmware updates to comply with IEC 62304.
- Continuously monitor risk through post-market surveillance and CAPA systems.
Insight: A structured risk management framework supports both regulatory approval and patient safety.
4. Ensure Data Security and Compliance
- For US markets, ensure HIPAA compliance, including secure hosting, encryption, audit logging, and role-based access.
- For EU markets, comply with GDPR while managing clinical data and PHI.
- Separate development and production environments to prevent unauthorized access to sensitive data.
- Encrypt data in transit and at rest, and implement secure authentication (2FA, session timeouts).
Example: Mobile medical apps should anonymize PHI when using development or test environments.
5. Adopt Agile Practices Mindfully
- Agile development accelerates delivery but must maintain traceability, documentation, and risk records.
- Follow AAMI TIR45 guidance for regulated software, linking user stories, tests, and risk mitigation.
- Conduct internal audits during each sprint to ensure compliance is maintained.
Insight: Agile does not conflict with compliance – structured processes make it possible to be fast and regulatory-ready.
6. Strengthen CAPA and Quality Management
- Implement a centralized CAPA system to track corrective and preventive actions.
- Perform root cause analysis and document preventive measures.
- Integrate CAPA into both FDA QSR and EU MDR quality systems.
Tip: Use CAPA findings to continuously improve development processes, risk controls, and documentation practices.
7. Prepare for Audits and Inspections
- Conduct regular internal audits of design, production, risk management, and post-market surveillance.
- Ensure that documentation is verifiable and accessible.
- Simulate FDA and EU MDR inspections to identify gaps before actual audits.
Insight: Proactive audit readiness reduces regulatory risk and builds confidence with authorities.
8. Plan for Post-Market Surveillance
- Develop post-market surveillance plans that comply with FDA and EU MDR requirements.
- Monitor device performance, user feedback, and adverse events.
Feed data back into risk management and CAPA processes to improve product safety and compliance continuously.
Strategic Cost Analysis: Budget Planning for Market Entry
Developing and launching a medical device requires not only technical precision and regulatory alignment but also strategic budget planning. The cost of market entry in the US vs. EU varies significantly due to differences in approval processes, documentation requirements, and post-market surveillance obligations. A proactive financial strategy can prevent delays and reduce the hidden cost of firmware bugs, compliance rework, and certification setbacks.
Cost Drivers in Regulatory Compliance
| Cost Category | EU MDR Market Entry | FDA Market Entry |
| Regulatory Submission | Costs for Notified Body assessment, conformity testing, and CE marking (varies €30K–€200K). | 510(k) submission or PMA fees ($20K–$400K+ depending on class). |
| Clinical Evaluation | Extensive documentation and clinical evidence required for MDR; often adds 6–12 months. | Clinical studies required only for Class IIb–III or novel devices. |
| Quality Management | ISO 13485 certification mandatory. | QSR compliance under 21 CFR Part 820 required (alignment with ISO 13485 underway). |
| Post-Market Surveillance | Ongoing vigilance, incident reporting, and regular updates to the EU database (EUDAMED). | Periodic post-market reports and FDA inspections. |
| Data Security & Privacy | Compliance with GDPR and cybersecurity regulations. | Compliance with HIPAA and cybersecurity guidance for connected devices. |
Insight: The EU’s MDR demands deeper documentation and clinical evaluation, while the FDA process often requires more extensive premarket testing and inspections. Both frameworks reward early planning and clear evidence management.
Hidden Costs and Risk Mitigation
Many development teams underestimate hidden regulatory costs, including:
- Revisions after failed audits or incomplete documentation
- Firmware vulnerabilities leading to re-certification delays
- Repeated submissions due to unclear risk assessment documentation
- Extended time-to-market from slow internal QMS adoption
At Developex, we help companies identify and mitigate these hidden costs early, combining technical and compliance expertise to optimize development budgets. Our teams perform regulatory impact assessments during product design, ensuring that all hardware, firmware, and software elements meet MDR and FDA expectations from the start.
By integrating quality management, risk control, and cybersecurity frameworks into development pipelines, Developex enables clients to:
- Shorten validation cycles
- Avoid hidden cost of firmware bugs and post-market recalls
- Achieve faster and more predictable market entry
Strategic Budgeting Tips for Regulatory Success
- Build a Dual-Market Roadmap – Align your design documentation and testing to meet both FDA and EU MDR standards simultaneously.
- Allocate a Contingency Fund (10–15%) – Cover potential compliance audits, device testing, or labeling changes.
- Invest in Traceability Systems – Use digital QMS tools for maintaining up-to-date risk assessments and design histories.
- Plan for Continuous Compliance – Include costs for post-market monitoring, CAPA processes, and periodic re-certifications.
- Partner with Experienced Development Teams – Collaborate with a partner like Developex who understands the intersection of medical software engineering and regulatory compliance.
Budgeting for regulatory compliance is not just an operational necessity – it’s a strategic investment in long-term product success. Companies that plan for both technical excellence and regulatory readiness from the outset enter markets faster, avoid costly delays, and build stronger relationships with regulators.
Final Thoughts
Navigating the regulatory landscape for medical devices in both the US and EU markets is complex but achievable with a strategic, compliance-first approach. Success depends on understanding the distinct requirements of the FDA, EU MDR, and related standards such as ISO 13485, ISO 14971, and IEC 62304, while maintaining rigorous HIPAA and GDPR compliance for patient data.
Medical device development teams that adopt a proactive, harmonized approach – combining FDA and EU MDR requirements, ISO standards, Agile methodologies, and robust data security – can:
- Minimize regulatory risk
- Accelerate time-to-market
- Maintain high-quality standards
- Build trust with regulators, clinicians, and patients
By understanding the key differences between US and EU regulations, applying international standards, and implementing practical compliance strategies, teams can ensure their devices are safe, effective, and globally market-ready.
Ensure your device meets every regulatory requirement before launch. Partner with Developex – our team ensures your medical software and embedded systems meet all regulatory, quality, and safety standards from design to launch.




